Technologies
Quantitative (comparative) proteomics

Quantitative mass spectrometry (MS) is a powerful analytical technique used to measure the relative abundance of proteins or other molecules in complex samples. Labeled quantitative MS methods involve the introduction of stable isotopic labels into samples, either chemically or metabolically, which allows for accurate quantitation and comparison of different samples. SILAC (e.g., Smolka lab) and TMT (e.g., Yu Lab) are two quantitative approaches that are well established and frequently used. Labeled Free Quantification (LFQ) methods, on the other hand, rely on the relative abundance of peptides in different samples, typically based on their ion intensities, to quantify changes in protein abundance. With recent rapid development of Data-Independent-Acquisition (DIA) workflows, LFQ-DIA (e.g., Yu Lab) methods have shown promise with much improved coverage and accuracy.
Post-translational modifications (PTM)
Post-translational modifications (PTMs) are essential for many biological processes, including protein folding, stability, localization, and activity. They are also frequently dysregulated in human diseases, however, their complexity and dynamic nature make them difficult to study using traditional biochemical methods. PTM analysis by MS has become an essential tool for many areas of biological and biomedical research.

Phosphoproteomics involves the fragmentation of phosphorylated proteins into peptides, followed by their separation and detection by mass spectrometry. By analyzing the mass and fragmentation patterns of these phosphopeptides, specific phosphorylation sites and changes in their abundance in response to different stimuli or conditions can be quantified (e.g., Smolka Lab). This information can provide important insights into the functional roles of phosphorylation in health and disease and can help guide the development of new therapies for a wide range of conditions.

Glycoproteomics: protein glycosylation is a common post-translational modification that plays important roles in cellular processes such as cell signaling, adhesion, and differentiation. Glycoproteins are digested into glycopeptides and this requires specialized sample preparation techniques that preserve glycan structures, such as enzymatic release and purification of glycopeptides, and derivatization to stabilize labile glycan moieties (e.g., DeLisa Lab). Glycoproteomics involves identification of glycan structures and glycosylation sites on glycoproteins. Quantitative glycoproteomics mass spectrometry can be achieved using either label-free or stable isotope labeling methods, which enable the comparison of glycosylation patterns and site occupancy between different samples. The study of the glycoproteome using mass spectrometry has advanced our understanding of the roles of glycosylation in cellular physiology and disease, and has enabled the development of new diagnostic and therapeutic strategies based on targeting glycoprotein biomarkers.
Protein-protein interaction & protein complex analysis
Mass spectrometry is a versatile and sensitive analytical tool to study protein-protein interactions and protein complex formation. One popular approach is affinity purification followed by mass spectrometry (AP-MS), which involves the isolation of a protein or protein complex of interest using an affinity tag, followed by MS-based identification of interacting proteins. AP-MS can identify both stable and transient interactions and can provide insights into the composition, stoichiometry, and dynamics of protein complexes (e.g., Yu Lab and Schang Lab).
Crosslinking mass spectrometry and structural proteomics

 Cross-linking MS (XL-MS) employs chemical cross-linkers to stabilize protein-protein interactions prior to MS analysis, which allows for the detection of more transient interactions, under native-like conditions. These approaches can provide complementary information to AP-MS and can help to validate and refine interaction networks. Recently, a new-generation of cross-linkers were developed (e.g., DSSO), which encompasses MS-labile bonds that yield fragment ions with a signature mass difference in the MS2 spectrum and enable peptide identification in the MS3 spectrum. These new cross-linkers enables a MS2-MS3 XL-MS workflow that minimizes the database search space from n2 to 2n and reduces the number of potential false positives to overcome limitations of the standard “MS2-centric” cross-link search programs. The Yu Lab designed a novel “MS3-centric” search engine for crosslink identification, called MaXLinker, with superior sensitivity, specificity and flexibility. Yu and Lin labs are working together to develop enrichable MS-cleavable cross-linkers to further improve the performance of XL-MS.
Cross-linking MS (XL-MS) employs chemical cross-linkers to stabilize protein-protein interactions prior to MS analysis, which allows for the detection of more transient interactions, under native-like conditions. These approaches can provide complementary information to AP-MS and can help to validate and refine interaction networks. Recently, a new-generation of cross-linkers were developed (e.g., DSSO), which encompasses MS-labile bonds that yield fragment ions with a signature mass difference in the MS2 spectrum and enable peptide identification in the MS3 spectrum. These new cross-linkers enables a MS2-MS3 XL-MS workflow that minimizes the database search space from n2 to 2n and reduces the number of potential false positives to overcome limitations of the standard “MS2-centric” cross-link search programs. The Yu Lab designed a novel “MS3-centric” search engine for crosslink identification, called MaXLinker, with superior sensitivity, specificity and flexibility. Yu and Lin labs are working together to develop enrichable MS-cleavable cross-linkers to further improve the performance of XL-MS.
Intact protein analysis
Intact protein analysis (e.g. Lin Lab) is popular due to its sensitivity and ability to analyze proteins in solution. ESI-MS can provide information on intact protein mass, subunit stoichiometry, and the presence of post-translational modifications such as phosphorylation and glycosylation. MALDI-MS is another commonly used technique for intact protein analysis, particularly for larger proteins and protein complexes, due to its ability to generate higher charge states and reduce fragmentation. SEC-MS can also be used for intact protein analysis, by separating intact proteins based on their size and shape prior to MS analysis. The combination of intact protein analysis with other MS techniques, such as bottom-up proteomics and cross-linking MS, can provide a more comprehensive understanding of protein structure, function, and interactions.
Mass Cytometry
Mass cytometry is a technology that combines flow cytometry with inductively coupled plasma mass spectrometry (ICP-MS) to enable high-dimensional, single-cell analysis. In mass cytometry, cells are labeled with isotopically pure metal-tagged antibodies, which can be detected by ICP-MS. This approach allows for the simultaneous detection of up to 50 parameters, including surface and intracellular markers, cytokines, and functional assays, at the single-cell level. Mass cytometry (Elemento Lab and Hiranmayi Ravichandran) has several advantages over traditional flow cytometry, including increased sensitivity, reduced spectral overlap, and the ability to detect rare cell populations. Additionally, mass cytometry enables the discovery of new cell subsets and biomarkers, as well as the identification of novel signaling pathways and cellular interactions. Despite its many advantages, mass cytometry has some limitations, including the requirement for specialized equipment, the high cost of reagents, and the complexity of data analysis.
Proximity-labeling MS
Proximity-labeling MS enables the identification of protein-protein interactions and the subcellular localization of proteins within cells. The technique involves the introduction of a biotin-containing molecule, such as biotin-phenol or biotin-tyramide, into cells, which is activated by a specific enzyme or protein of interest. The activated biotin then covalently labels nearby proteins, allowing them to be enriched and identified by MS.
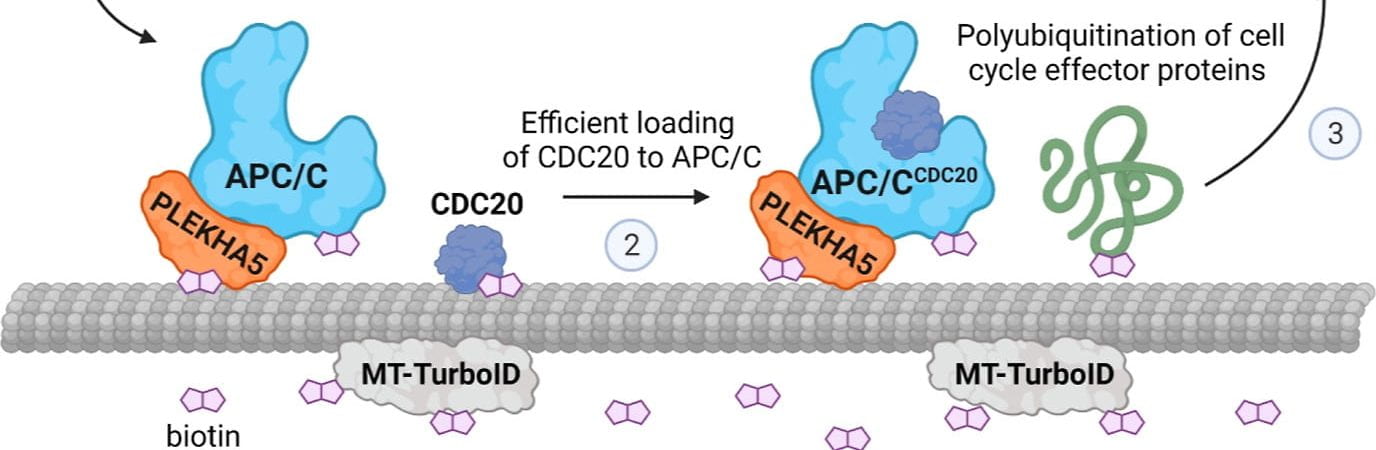
Labeling specific proteins: APEX2 and TurboID are two examples of proximity-labeling enzymes that have been developed for use in MS. These enzymes are fused to a protein of interest and expressed in cells, where they generate reactive intermediates that covalently label nearby proteins with biotin. The labeled proteins can then be enriched and identified by MS, enabling the identification of protein-protein interactions and subcellular localization of proteins. APEX-MS (e.g., Lis lab) and TurboID (e.g., Smolka Lab and Baskin Lab) have several advantages over other proximity-labeling techniques, including their high labeling efficiency, rapid kinetics, and specificity for labeling proteins in the immediate vicinity of the fusion protein. Additionally, these enzymes can be used in a variety of cell types and model organisms, and have been used to study a wide range of biological processes, including signaling pathways, protein complexes, and organelle proteomes. In summary, proximity-labeling MS, and specifically APEX-MS and TurboID, offer a powerful approach to the study of protein-protein interactions and subcellular localization, providing valuable insights into the organization and function of the proteome within cells.
Labeling specific genomic loci: Instead of tagging specific proteins with APEX2 or TurboID, CAPTURE (dCas9-BirA) or c-BERST (dCas9-APEX2) use dCas9 to bring enzymes to individual genomic loci to biotinylate factors associated with specific regulatory elements (such as enhancers, promoters, and others) to specifically map proteome microenvironment around these loci (Yu Lab in close collaboration with Apostolou Lab at WCM).
Single cell proteomics
Single-cell and spatial omics are revolutionizing biology; single-cell transcriptomics studies have clearly shown the heterogeneity of gene expression across cells and the importance of measurement at the single cell resolution. Proteins are the functional molecules in the cell, so it is naturally important to measure protein abundance and their changes at the whole proteome level across different cells with high spatial resolution (ideally at the single cell level). Complementing the existing antibody-based spatial/single-cell proteomics methods that employ imaging, barcoding, or mass spectrometry (e.g., CyTOF), obtaining a comprehensive view of the entire proteome at a high spatial resolution is crucial for uncovering novel factors that play a pivotal role in biological processes or diseases. With the recent development in mass spectrometry instrumentation (significantly increased sensitivity), multiplexing with tandem mass tags (TMT), Data-independent-acquisition (DIA) with deep neural network enabled search algorithms, and development of equipment for ultra low volume liquid and cell handing, innovative ultra-sensitive proteomics technologies with high spatial resolution are being developed (Yu Lab and De Vlaminck Lab).
Our Proteomics Facilities at both Ithaca and WCM in NYC provide excellent service for most of these technologies with robust performance. Individual labs in CIP also excel in developing specific technologies and applying them to answer biological questions.